A challenging picture
According to a study carried out in 2001 [1], 9.4% of women over forty (i.e., since a very young age) and 11.8% of men over sixty are affected by osteoporosis. The numbers increase considerably if referred to osteopenia (for the same population groups the figure is 47.2% for women and 46.1% for men), and even more so in proportion to the age of the patients’ sample considered. This figure points out a major problem, which is not easy to solve.
The guidelines [2,3] base their approach to this disease mainly on the intake of calcium and vitamin D, estrogens, bisphosphonates (for glucocorticoid-induced osteoporosis), but focus primarily on prevention at a younger age. Unfortunately, it is not uncommon for these therapeutic strategies to fail to achieve the desired results, mainly because of endocrine-metabolic conditions that limit their applicability, and even lead to a series of side effects due to the therapies themselves.
In order to identify the most effective therapeutic strategy, it is first of all necessary to understand the main reasons for the ongoing bone demineralization, given that they may be different and not be solved by a single therapy.
Understanding bone metabolism
The bone structure [4-7] is composed of highly mineralized connective tissue (mineral substance makes up about 70% of bone), and is crossed by the necessary blood and lymphatic vessels. Bone cells are also part of the organic substance in the bone, which is mostly type 1 collagen, and are involved in the various stages of bone metabolism. These cells fall into two basic categories:
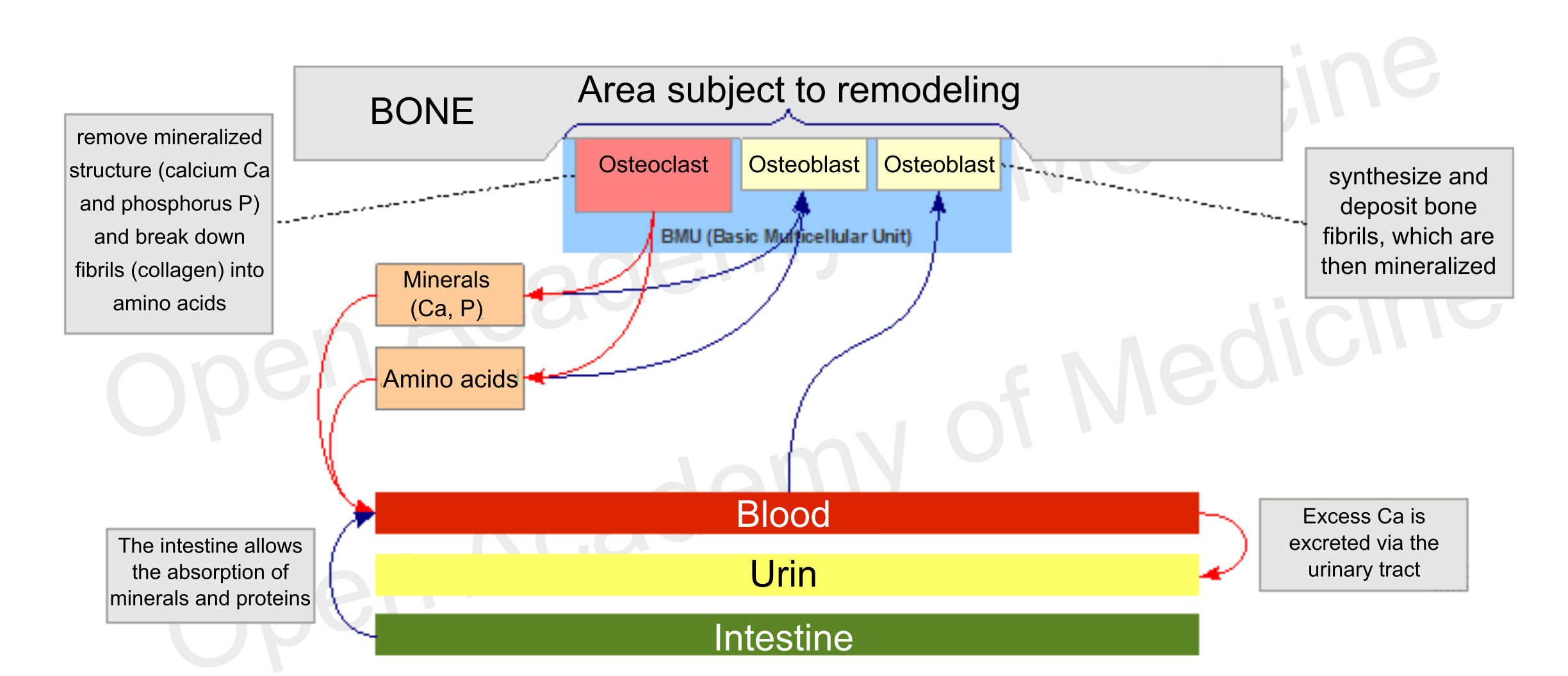
Figure 1: main interactions involved in bone metabolism
These cells do not act on their own, they move in groups called BMUs (Basic Multicellular Units), consisting essentially of a front of osteoclasts followed by a certain number of osteoblasts. The prevalence of the activity of one of these two types of cells is the main driver for differentiating the growth, maintenance (remodeling) and demineralization phases of the skeleton. The maximum level of bone density is usually reached around twenty years of age, while after thirty it generally starts to decrease.
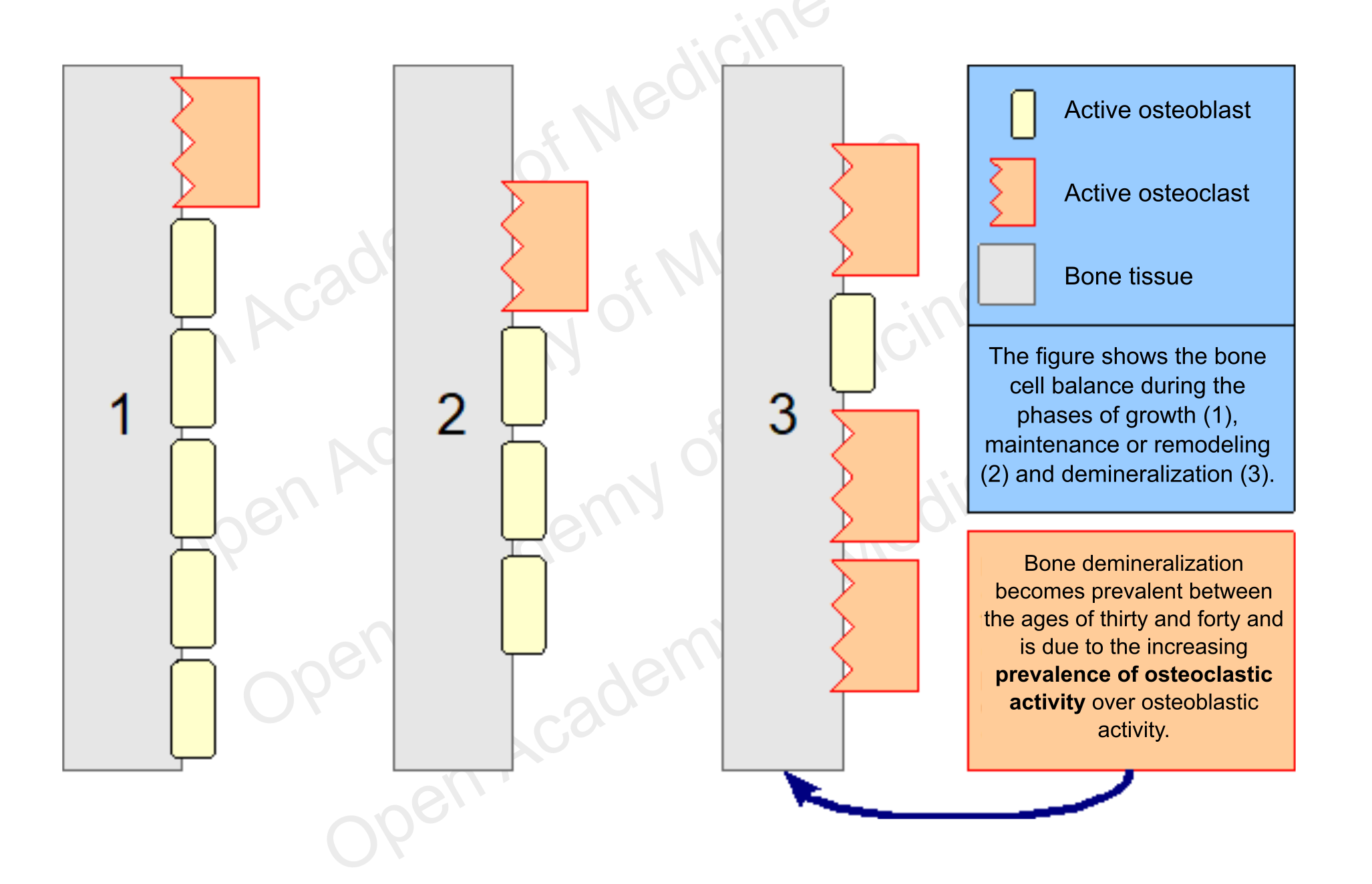
Figure 2: schematic representation of bone cell balancing
The balance of these antagonistic processes depends on several factors that can influence the time of cell apoptosis (the life span of osteoclasts is generally much shorter than that of osteoblasts) or the formation of new osteoblasts and osteoclasts.

Figure 3: BMU activation
During their bone remodeling activity, osteoblasts can get stuck in the matrix formed by themselves, thus evolving into osteocytes: these are cells that, through long cytoplasmic membrane outgrowths, form an exchange network running through the bone tissue which can communicate with both the bone marrow and the BMUs (e.g. to stimulate remodeling of a particular bone fragment).
The bone tissue that does not undergo remodeling is generally protected by a micro-layer of collagen, surrounded by a layer of bone lining cells (i.e., cells derived from inactivated osteoblasts): this organic material prevents osteoclasts from adhering to the mineralized surface and is possibly removed by the lining cells which, through the mediation of osteocytes, can be stimulated to produce collagenase (which degrades type 1 and 2 collagen).
Changes in bone density
The decrease in bone density is a process that begins between thirty and forty years of age in both men and women, due to an increasing disproportion in the number of processes involved in bone tissue regeneration that are characterized by higher osteoclastic activity. In most cases this development is not related to a low calcium intake, but rather to other factors, mainly endocrine and metabolic ones, which can influence osteoblast and osteoclast activity. However, it is the predominance of osteoclasts over osteoblasts that leads to gradual bone demineralization (and consequently to bone fragility).
Endocrine factors
Among the factors that are most commonly involved in the imbalance between the action of osteoclasts and osteoblasts – in addition to sex hormone (estrogen, testosterone) deficiency – is a chronic excess of glucocorticoids [8-15] (both endogenous and exogenous; BIA-ACC - Flat Low/High HPA axis index).
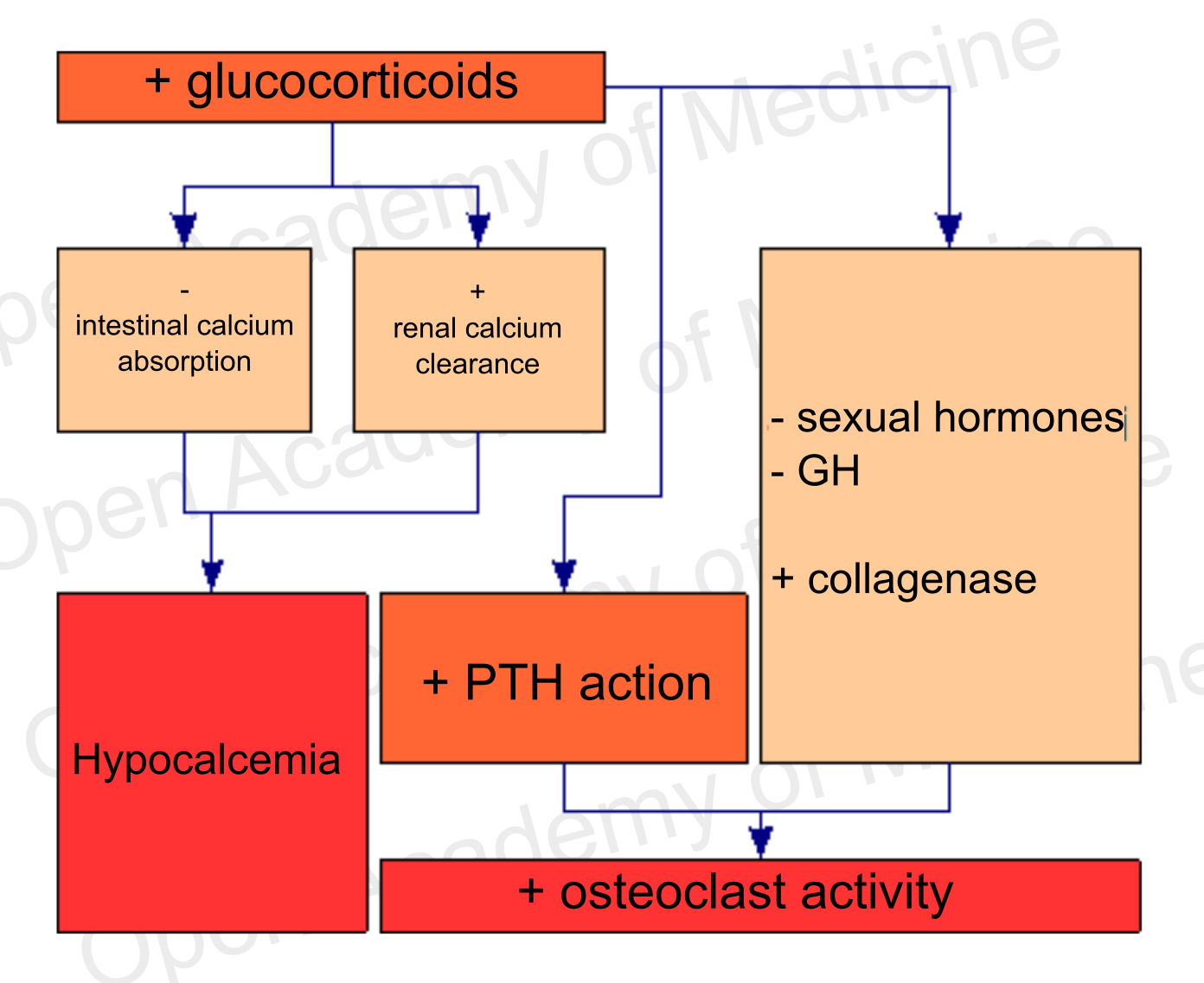
Figure 4: glucocorticoids and bone metabolism (BIA-ACC - Flat Low/High HPA axis index)
There is evidence of the effects that this excess triggers on bone metabolism, through mechanisms that are both direct on bone cells and mediated by local and systemic interactions with hormones, growth factors and cytokines.
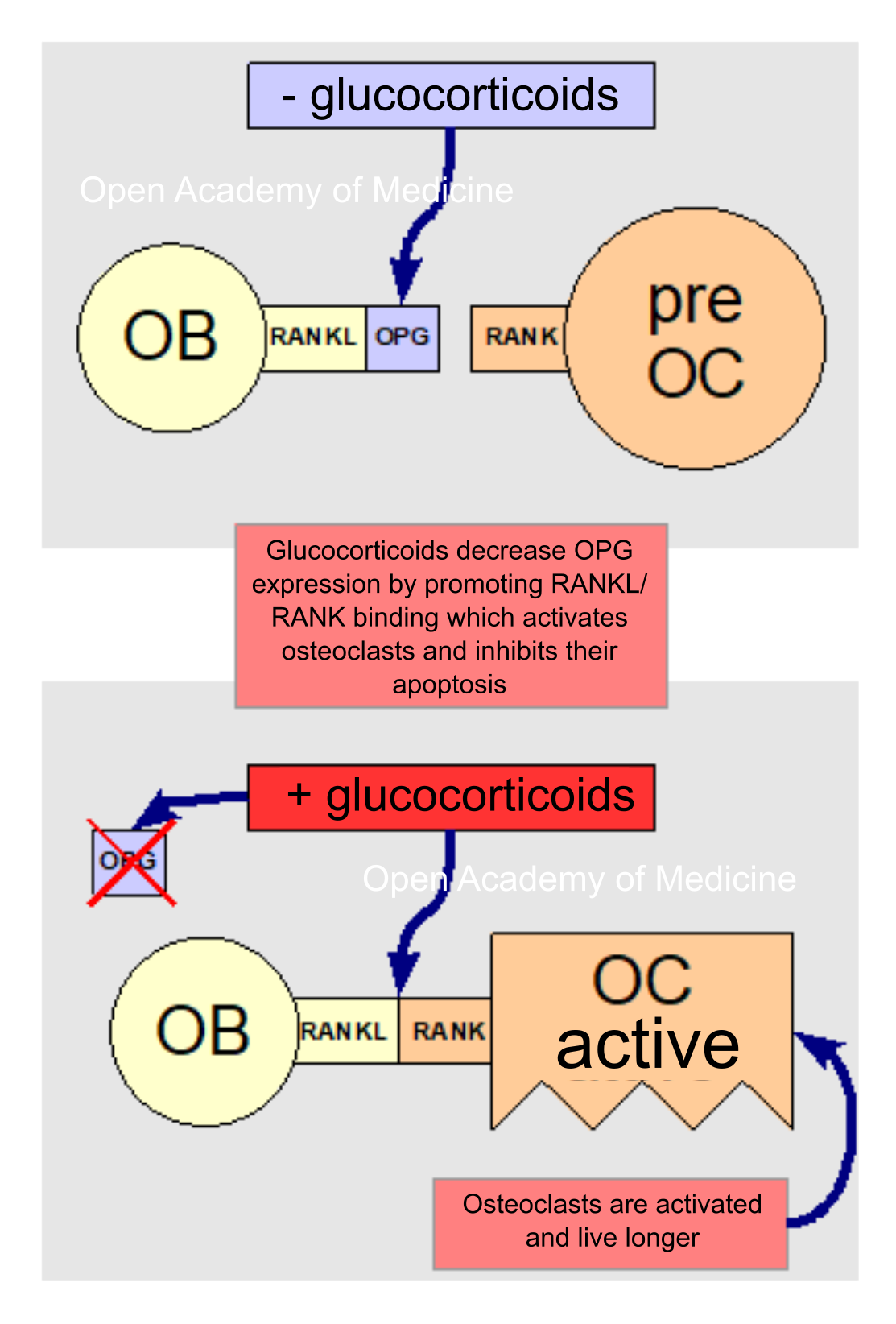
Figura 5: glucocorticoids and RANKL/OPG/RANK binding
At an endocrine level, glucocorticoids reduce intestinal calcium absorption while increasing renal calcium clearance, thus causing hypocalcemia. This condition is associated with increased osteoclast activation and hence to an increased bone resorption (research will have to clarify whether this is due to glucocorticoid-induced hyperparathyroidism or to increased PTH sensitivity by osteoblasts, which would react to this hormone by activating the osteoclasts). Glucocorticoids also inhibit GH secretion and sex hormone synthesis, while increased glucocorticoids are associated with an increased collagenase expression, that has negative effects on organic bone matter (see HPA axis for more details: Insomnia, mood disorders, anxiety, melancholy, depression, MUS, panic attacks).
Glucocorticoids also have direct effects on bone cells through the RANKL/OPG cytokine system [7,16] by increasing the expression of the former and decreasing that of the latter, and thus promoting the binding of RANKL cytokines (expressed on osteoblast membranes) to RANK receptors (on the membranes of osteoclast progenitors); this binding stimulates the differentiation and activation of osteoclasts and inhibits their apoptosis (programmed cell death).
The increase in glucocorticoids also inhibits the osteoblast differentiation factor, thus reducing the bone regeneration activity, and increases osteoblast and osteocyte apoptosis as well. Osteocyte apoptosis also plays a key role in bone fragility, since osteocytes – as already seen – are involved in BMU activation and bone remodeling processes.
The effects described above are due both to exogenous glucocorticoid excesses [9,14] (and should therefore be particularly taken into account in patients taking glucocorticoids for therapeutic purposes) and to endogenous ones. Among the factors that are involved in chronic excess cortisol and the related circadian rhythm shift (BIA-ACC - Flat Low/High HPA axis index), there are the presence of chronic inflammatory processes, chronic stress on the HPA axis and poor nutrition [26-29].
High-titer EPA+DHA omega-3 fatty acids have been shown to reduce the expression of RANKL cytokines and increase OPG [17-21], thereby decreasing osteoclast activation; high-titer EPA+DHA omega-3 fatty acids intake is also associated with a reduction in the concentration of proinflammatory cytokines (e.g. IL-6 and TNF-α), so the effects of these fatty acids contribute in several directions to reducing bone absorption. Recent studies [22,23] have also reported that certain hop flavonoids (Humulus Lupulus L.), in particular xanthohumol, are able to interact selectively with estrogen receptors and exert an estrogen-like activity on bone metabolism (thus reducing demineralization), without producing the side effects of hormone therapies (several publications report an increased risk of breast and uterine cancer), while also showing a protective effect against these problems (see high titration EPA+DHA+Humulus Lupulus L. supplementation).
Metabolic factors
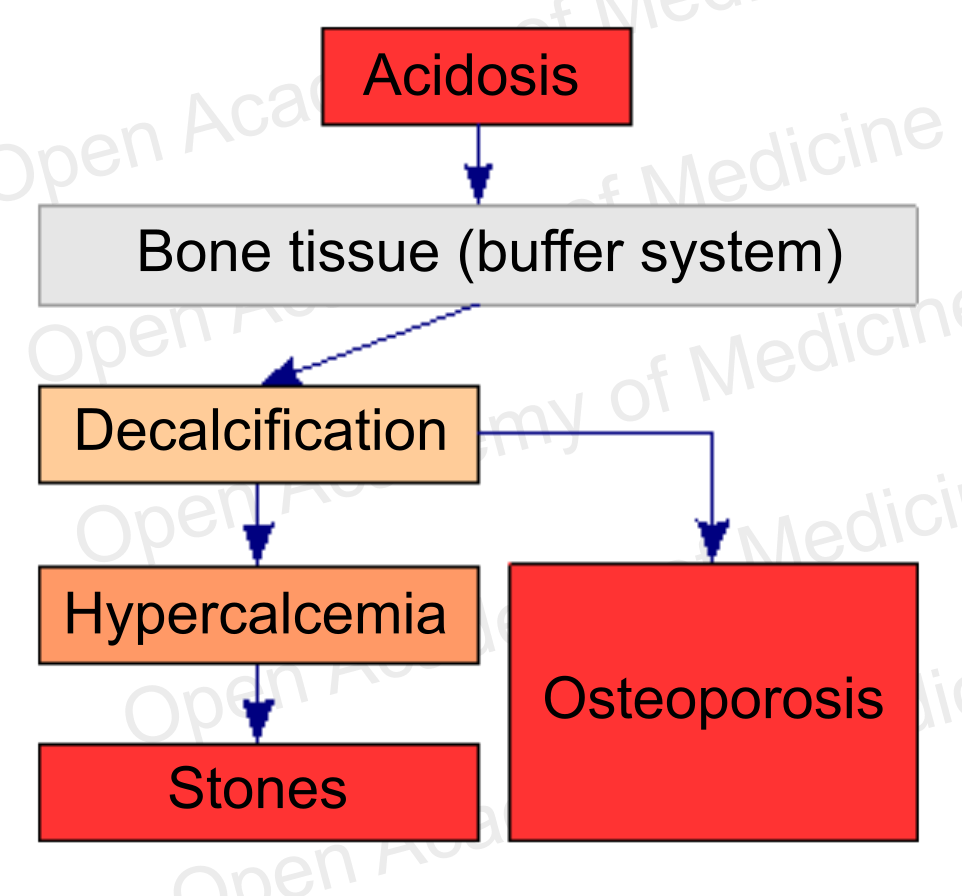
Figure 6: acidosis and bone metabolism (see supplementation with buffer systems)
Bone remodeling is not only instrumental in tissue regeneration and in the adaptation of the skeletal structure to the mechanical stresses to which it is subject, it is also an essential process for calcium homeostasis, as bone tissue is the body’s main calcium reservoir. However, the balance with free ionized calcium in the plasma (and its possible deficiency) is not the main aspect to be considered from this viewpoint: it is known that bone tissue plays a fundamental role in balancing body pH, as it is itself a buffer system against acidosis [24,25]. The decalcification produced to preserve body pH can obviously lead to increased bone fragility and hypercalcemia.
Therefore, from a therapeutic standpoint, in order to cope with such a situation it is necessary to take proper supplementation with phosphate and bicarbonate buffer systems (see supplementation with buffer systems): restoring the pH becomes a priority [30-31] over replenishing calcium, given that calcium may already have excessive plasma concentrations due to the ongoing bone demineralization.
Why should a therapy be instituted?
If the bone tissue is subject to demineralization for one or more of the aforementioned reasons, it will be necessary to treat the patient to correct this trend prior to considering the usual calcium and vitamin D supplementation (which may not be necessary), as the excess of these components, deprived of the systemic conditions for bone tissue recovery, would easily lead to side effects such as soft tissue calcifications or kidney stones, and would not be able to limit the ongoing bone mineral loss.
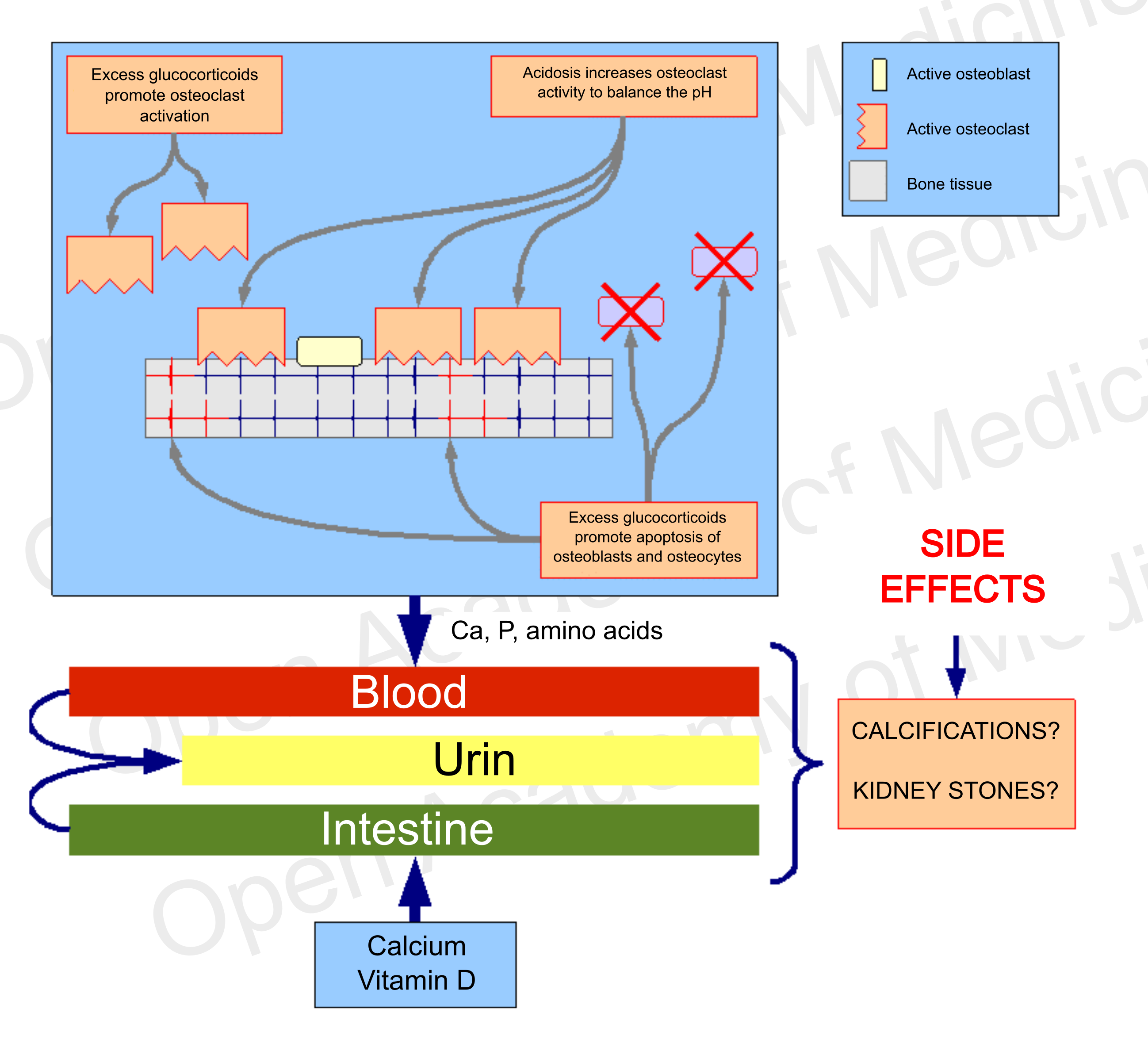
Figure 7: side effects of a therapy that does not address endocrine and metabolic aspects
Authors: Dario Boschiero - Date: 14/12/2020
Attention: these contents can be freely used for personal learning purposes only. The use is regulated by Law No. 633/1941 and subsequent amendments, as well as by the copyright and patent legislation in force. Any use for commercial and profit-making purposes is forbidden.
References




